التلاسيميا “فقر دم البحر المتوسط”: أنواع وأعراض وعلاج
1 min read
شكل (1): السخنة الخاصة بالتلاسيميا.
فحوصات ما قبل الزواج أهم وسيلة للوقاية من الإصابة بالتلاسيما (بيتّا) الكبرى
مقدمة:
بشكل عام، لكل صفة من صفات جسم الإنسان (لون الشعرأو العيون أو لون البشرة أو زمرة الدم ..الخ) هناك صبغي Gen واحد يتم توارث نصفه من الأب ونصفه من الأم، وهذا ينطبق على الأمراض الوراثية أيضا، ويجب أن يرث الطفل الصفة المرضية من كلا الوالدين حتى يظهر لديه المرض، هذا ينطبق على أمراض فقر الدم الوراثية التي سنتحدث عنها: التلاسيميا أو (فقر دم البحر المتوسط) في هذا الموضوع وفقر الدم المنجلي في الموضوع القادم، وذلك لأنهما الأكثر انتشاراً في منطقة الشرق الأوسط.
التلاسيميا Thalasaemia
هي خلل وراثي في تركيبة الهيموغلوبين (خضاب الدم). تختلف شدتها من الناحية السريرية بشكل كبير إذ تتراوح بين الأشكال البسيطة غير المصحوبة بأعراض إلى الأنواع الشديدة أو حتى المميتة.
سبب تسمية هذا المرض بـ “تلاسيميا” أو ما يعرف بفقر دم البحر الأبيض المتوسط، يعود لليونانيين الذين كانوا يعيشون قرب البحر المتوسط وكان بعضهم يعاني من هذا المرض، إذ تتألف الكلمة من قسمين، الأول “تالاسا” والتي تعني البحر، “هايما” والتي تعني الدم. لكن يمكن أن يتواجد هذا المرض في العديد من المناطق في العالم
قبل البدء بالحديث عن التلاسيميا، لابد من ذكر بعض المعلومات الضرورية لفهم المرض.
تصيب التلاسيميا الهميوغلوبين (خضاب الدم) وهو الجزء من الكريات الحمراء المسؤول عن نقل الأكسجين إلى جميع أنحاء الجسم.
يتكون الهميوغلوبين من 4 سلاسل من البروتين (اثنتان من ألفا-غلوبين واثنتان من (بيتّا)-غلوبين) ويتوضع ضمن هؤلاء الجزيئات الأربعة جزيئه الـ “هيم”، عند وجود مشكلة في تشكُّل أي من هذين النوعين من الغلوبين (ألفا أو (بيتّا) تحدث التلاسيميا. التلاسيما (ألفا) تحدث عند وجود مشكلة بالسلسلة ألفا، التلاسيميا (بيتّا) تحدث عند وجود مشكلة بالسلسلة (بيتّا). تعتبر التلاسيما (بيتّا) الأكثر انتشارا، لذلك سيتم التكلم عنها باستفاضة.
التلاسيميا (بيتّا)
تقسم التلاسيميا (بيتّا) إلى ثلاثة أشكال حسب شدة الحالة السريرية وهي:
- الثلاسيميا (بيتّا) الكبرى.
- الثلاسيميا (بيتّا) المتوسطة
- الثلاسيميا (بيتّا) الصغرى. ويكون الفرد المصاب بها (حامل للمرض).
1- التلاسيميا (بيتّا) الكبرى:
يحدث هذا المرض عندما يكون لدى الشخص صفتان وراثيتان مصابتان ورثهما من الأب والأم، وبالتالي لا تتشكل السلسلة (بيتّا) من الهيموغلوبين، مما يؤثر على بنية الكريات الحمراء وقدرتها على نقل الاكسجين وكذلك على عمرها.
تحدث التلاسيميا (بيتّا) عند وزاج شخصين حاملين للمرض من بعضهما، أو زواج شخص مصاب بالتلاسميا (بيتّا) مع حامل للتلاسيميا (بيتّا)
تعد هذه الحالة خطيرة، حيث لا يمكن للجسم أن ينتج ما يكفي من الهيموغلوبين الصحيح أو الكريات الحمراء السليمة لذلك سوف يحتاج المريض للعلاج طوال حياته.
تبدأ الأعراض بالظهور من عمر3-6 أشهر من الولادة وتشمل شحوباً في الوجه وضيقاً في التنفس ويرقان (تلون الجلد وبياض العين باللون الأصفر).
فيما بعد، وبسبب محاولة نقي العظم لتصنيع المزيد من الكريات الحمراء للتعويض عن فقر الدم الحاصة وتخرب الكريات الحمر بسرعة، تحدث الأعراض التالية:
الأعراض الناجمة عن نقص تشكل الهيموغلوبين السليم:
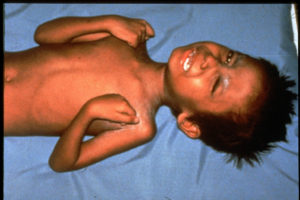
- مشاكل بنمو العظم، وهذا يؤدي لضخامة بعظام الوجه ويعطي الطفل أو المصاب سحنة وملامح خاصة (الشكل 1).

شكل (1): السحنة الخاصة بالتلاسيميا. - مشاكل بالنمو، مما يسبب قصر القامة وضآلة الجسم.
- ضخامة طحال.
- تقرحات بالساق.
- زيادة التأهب للالتهابات والعدوى بالأمراض.
- تأخر سن البلوغ.
- انقطاع الطمث مبكراً.
- قد تحدث إصابة بالسكري أو هشاشة العظام.
- احتمال الوفاة في مرحلة الطفولة المبكرة.
الأعراض الناجمة عن تراكم الحديد في الدم والجسم:
يحدث ذلك بسبب نقل الدم المتكرر لعلاج فقر الدم الحاصل نتيجة تخرب الكرايات الحمراء بسرعة، مما يؤدي لانطلاق الحديد الذي يدخل بتركيب الهيموغلوبين.
تشمل هذه الأعراض:
- تعب شديد.
- التهابات متكررة
- ترقق العظام.
- الإصابة بالداء السكرى.
- تلون الجسم باللون الترابي.
- التهاب المفاصل.
- أضرار تصيب القلب والكبد نتيجة لتركم الحديد في النسج المكونة لهم.
2- التلاسميا (بيتّا) المتوسطة:
يمكن أن يعاني المريض المصاب بالثلاسيميا (بيتّا) المتوسطة من أعراض مماثلة للثلاسيميا (بيتّا) الكبرى ولكنها تكون أقل شدة، قد تسوء الحالة مع الوقت، وقد يحتاج في مرحلة ما إلى نقل الدم بصورة منتظمة في بعض اللأحيان.
الأسباب المؤكدة لعدم ظهور الأعراض الشديدة على مرضى التلاسيميا (بيتّا) المتوسطة بشكل مماثل لما يحصل في الحالة الكبرى غير مفهوم، ولكن لسبب ما يستطيع الشخص المصاب تكوين كمية من الهيموغلوبين كافية لحاجاته دون الحاجة لعلاج مكثف كما في حالات التلاسيميا الكبرى، وبعض التفسيرات لذلك هي:
- تمكن الجينات الوراثية المصابة من العمل بشكل جيد وإنتاج هيموغلوبين جيد وقادر على حمل الاكسجين بشكل كاف ولا تسبب تشوه بالكريات الحمراء وتخربها بسرعة.
- أو أن المورثات الأخرى السلمية تعمل مع المورثات المتضررة وتساعدها على العمل بشكل أفضل وإنتاج كمية الهيموغلوبين السليم.
- من الممكن أن يكون السبب هو اشتراك التلاسيما مع أمراض هيموغلوبين أخرى مثل
تلاسيميا (بيتّا) + هيموجلوبين E: هو منتشر في كمبوديا وتايلاند وأجزاء من الهند. تكون أعراض هذا المرض مشابهة من الناحية السريرية لتلاسيميا (بيتّا) الكبرى. تلاسيميا (بيتّا) + الهيموجلوبين المنجلي Sickle cell: : وهو منتشر في إفريقيا وسكان البحر الأبيض المتوسط، ويشابه من الناحية السريرية مرض فقر الدم المنجلي مع علامة إضافية تتمثل في تضخم الطحال.تلاسيميا (بيتّا) + الهيموجلوبين C : ينتشر لدى سكان البحر الأبيض المتوسط وإفريقيا، وتؤدي إلى تحلل الدم متوسط الشدة بالإضافة لتضخم الطحال .
3- التلاسيميا (بيتّا) الصغرى :
تحدث هذه الحالة في حال وجود مورثة واحدة فقط لدى الشخص تحتوي على خلل بإنتاج الهميوغلوبين (بيتّا) ولكن يوجد مورثة أخرى سليمة تماما، وبالتالي غالباً لا يتظاهر المرض بأي أعراض ويكون الشخص حاملاً للمرض وقد يتم كشفه فقط عند أجراء تحاليل دموية ورحلان للهيموغلوبين (Electrophoresis).
علاج التلاسيميا (بيتّا)
في حالة الثلاسيميا (بيتّا) الكبرى والحالات الحادة من الثلاسيميا (بيتّا) المتوسطة تحتاج علاجاً طوال الحياة.
- نقل الدم: يعتبر العنصر الأساسي للعلاج وقد يصل تواتره لمرة كل 4-8 أسابيع.
- التخلص من الحديد الزائد (الاستخلاب chelation therapy) ويتم ذلك باستخدام الأدوية الخاصة التي ترتبط بالحديد الزائد وتتخلص منه عن طريق البراز أو البول.
- اتباع نظام حياة صحي وحمية صحية متوازنة وتجنب الأطعمة الغنية بالحديد وممارسة التمارين بشكل منتظم وعدم التدخين أو تناول الكحول.
- استئصال الطحال: في حال تضخم الطحال، وذلك للتقليل من كمية الدم المنقولة بشكل متكرر نتيجة لتجمع الدم في الطحال المتضخم.
الشفاء التام يكون عن طريق عملية زراعة الخلايا الجذعية ويتم ذلك بنقل خلايا النخاع العظمي من متبرع سليم (غالبا يكون أحد الأخوة) إلى الشخص المصاب بالثلاسيميا (بيتّا) أو نقل الخلايا الجذعية من الحبل السري.
زرع نقي العظم يمكن أن يكون الخيار لبعض الناس ولكن في بعض الحالات قد تكون المخاطر أكبر من الفوائد المتوقعة.
إنذار مرض التلاسيميا (بيتّا)
يمكن تدبير بعض المشاكل الصحية الرئيسية التي تترافق مع فقر دم البحر المتوسّط عن طريق العلاج عادة ولكن تبقى هذه الحالة خطيرة وتؤثر بشكل كبير على نوعية الحياة وجودتها.
تحتاج التلاسيميا لمراقبة دقيقة وعلاج منتظم وإلا سيواجه الشخص زيادة في خطر أن تسبّب الأنواع الشديدة منها ضرراً للأعضاء وتهديداً للحياة.
في الماضي، كانت الحالات الشديدة لفقر دم البحر المتوسط تقضي على حياة الشخص في بداية البلوغ، ولكن اختلف الأمر حالياً مع ابتكار طرق جديدة للعلاج، حيث يأمل الأطباء أن يصل متوسط عمر المصابين بالتلاسيما إلى الخمسينات أو الستينات.
التلاسيميا (ألفا)
يوجد أربع مورثات تؤثر على تشكل هيموغلوبين (ألفا)، ويتطلب وجود ثلاث مورثات مصابة حتى نقول أن يتظاهر المرض، لذلك هو أقل انتشاراً من التلاسيميا (بيتّا).
هناك أربع أشكال سريرية لهذا المرض وذلك بحسب عدد الموروثات التي تعرضت للخلل:
- إصابة مورثة واحدة: تسمى ثلاسيميا ألفا الصامتة ويسمى الشخص المصاب بالحامل الصامت، لا يتم كشفها الا خلال فحوصات مخبرية خاصة.
- إصابة مورثتين: يكون لدى المصاب في هذه الحالة فقر دم معتدل، وتكون الكريات الحمراء صغيرة. يتم كشف هذه الحالة بفحص الدم الروتيني.
- إصابة ثلاث مورثات: يحدث في هذه الحالة فقر دم شديد ويتطلب نقل دم. يتشكل في هذه الحالة هيموغلوبين شاذ يدعى هيموغلوبين H وذلك نتيجة عدم تشكل هيموغلوبين طبيعي.
- إصابة أربع مورثات: تدعى هذه الحالة استسقاء الجنين hydrops fetalis. يتشكل في هذه الحالة هيموغلوين شاذ آخر يدعى هيموغلوين (بارت). يموت معظم المصابين بهذه الحالة أثناء الحمل أو بعد الولادة بوقت قصير.
الوقاية:
أهم خطوة للوقاية من التلاسيميا (بيتّا) الكبرى هي إجراء فحوصات ما قبل الزواج وذلك للكشف عن الحالات التي يكون بها الشخص حاملا للتلاسيميا، حيث أن زواج شخصين حاملين للتلاسيميا (بيتّا) تحمل خطر ولادة طفل لديه تلاسيميا (بيتّا) الكبرى بنسبة 25%.
المصادر:
https://www.nhs.uk/conditions/thalassaemia/




need cash now payday poor credit loans credit loans guaranteed approval